Phylogenetic Analysis of BC Data
Alan Selewa
2017-11-17
Load processed data
We load the familiar data structure for single-cell mutation profiles.
source("/Users/aselewa/sc_call_variants/readcounts_parser.R")
covmat <- gen.CovMat()Mutation Matrix
For each cell-mutation, we count the total number of reference and alternative alleles. Under the null model, the probability of observing n alternative alleles and N-n reference alleles is given by:
\[ P(A = n) = {{N}\choose{n}} p_{error}^{n} (1 - p_{error})^{(N-n)} \]
where \(p_{error} = 0.004\) is the estimated error rate. The error rate is define as the probability of observing a mutation by chance. We perform a binomial test to obtain the p-value of our observed alternative/reference counts under the null model. We choose a cut-off of 0.05. If a cell-mutation satisfies the cutoff, then a “1” is placed in the mutation matrix and 0 otherwise. Mutations with missing data are left as “NA”.
The mutation matrix was obtained for all mutation-cell pairs. Below we show the top 5 mutations in terms of minimal missing data.
phylomat <- gen.PhyMat(covmat)
NAcount <- apply(phylomat,1,function(x) length(which(is.na(x))))
subphylo <- t(phylomat[order(NAcount)[1:5],]) #get top 5 informative mutations
NAcount <- apply(subphylo,1,function(x) length(which(is.na(x))))
subphylo[order(NAcount),] #order cells by how informative their mutations are [,1] [,2] [,3] [,4] [,5]
BC03_13 1 0 0 1 1
BC03_28 0 0 1 1 0
BC03_50 0 0 1 1 0
BC03_58 0 0 1 1 0
BC03_09 0 1 1 NA 0
BC03_11 1 1 NA 1 0
BC03_16 0 0 1 NA 0
BC03_19 1 1 1 NA 1
BC03_20 1 1 NA 1 0
BC03_26 0 0 1 NA 0
BC03_29 1 0 1 NA 0
BC03_41 0 0 1 NA 0
BC03_43 1 1 NA 1 1
BC03_57 0 NA 1 1 0
BC03_78 1 0 1 1 NA
BC03_92 0 0 0 1 NA
BC03_94 0 1 1 1 NA
BC03_85 0 0 1 1 NA
BC03_17 0 1 NA NA 1
BC03_21 0 NA NA 1 0
BC03_23 1 0 1 NA NA
BC03_24 NA 0 NA 1 0
BC03_06 NA NA NA 1 0
BC03_35 NA NA 1 1 NA
BC03_60 NA NA 1 1 NA
BC03_81 0 0 NA NA NA
BC03_86 0 NA NA 1 NA
BC03_93 0 NA 1 NA NA
BC03_66 0 1 NA NA NA
BC03_03 NA NA NA 1 NA
BC03_37 NA 0 NA NA NA
BC03_53 0 NA NA NA NA
BC03_25 NA NA NA NA NAA lot of missing data. How do we deal with this? We can impute or try a method that does phylgenetic reconstruction with missing data, although that is probably not going to be a good tree.
CNV Profiles
It is difficult to distinguish copy-number variation from differential expression in scRNA-seq data. Instead, researchers thus far detect large-scale CNVs by ordering genes by their genomic position and performing a moving average over ~50-200 gene blocks, depending on the number of genes used in the analysis.
This idea was executed in Patel et al. and Chung et al. using single-cell RNA-seq data. We adopt this idea to try to infer large-scale CNVs.
As a reference, we use GTEx normal breast tissue TPMs. We average across all breast tissue. The single-cell data is centered using this averages, thus the final matrix is a relative expression matrix to GTEx. The relative expression matrix is then smoothed.
The CNV profiles are shown below. The black vertical bars seperate chromosomes. The order of chromosomes is from left to right as 1, 2, 3, .. ,X, Y. I used all the genes that occur in both GTEx and the single-cell data (~10k genes), with a gene block of 150 genes. The dendrogam on the left represents heirarichal clustering of the expression profiles.
source('/Users/aselewa/sc_call_variants/inferCNV.R')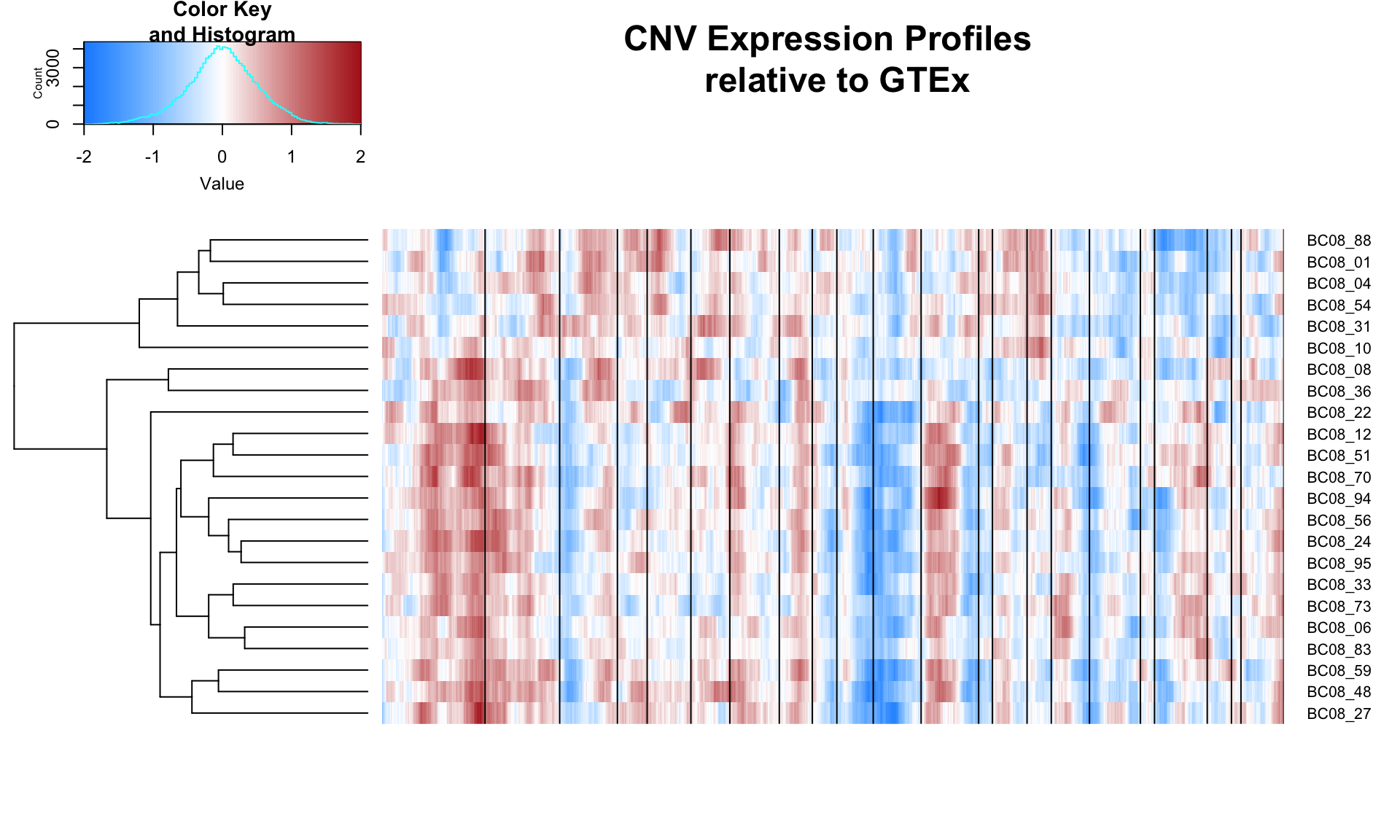
We have complete phylogeny for 4 cells in the mutation matrix, namely, BC03_13, BC03_28, BC03_50, and BC03_58. According to their mutation profiles, 13 is distant from 28,50,and 58, while these 3 cells have 0 distance between them. Does this pattern in the heirarchical clustering of CNV profiles hold? Yes, the trend holds.
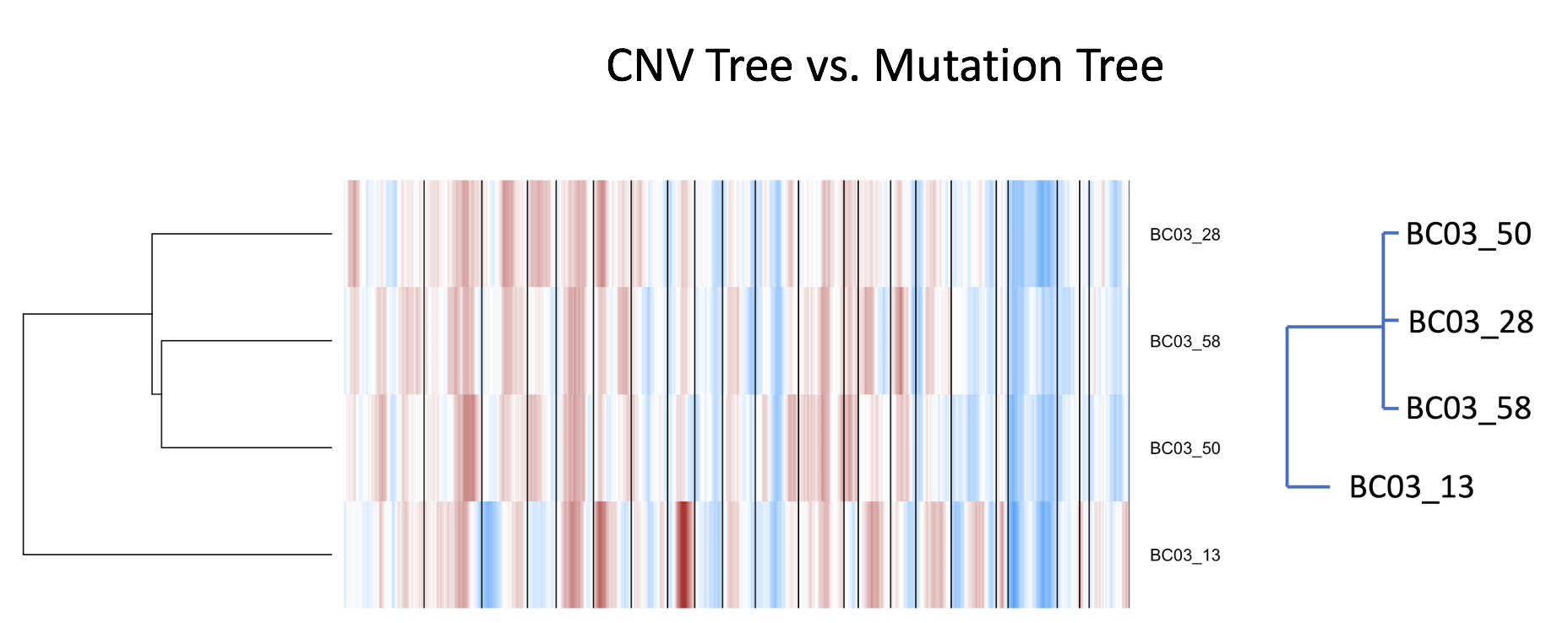
The figure above compares the two trees. The left tree is a little finer due to the nature of the data, but overall the trend holds. Perhaps we can use this CNV data to infer missing data in the original phylogenetic reconstruction problem.
For instance, BC03_57 has the following mutational profile:
subphylo["BC03_57",][1] 0 NA 1 1 0From the CNV profiles, BC03_57 shares a more recent ancestor with BC03_50, BC03_28, and BC058, thus the missing data is likely to be ‘1’, because the CNV tree tells us it is further from BC03_13 than BC03_58/28/50.