Heart Atlas - Donor 03231
Alan Selewa
Introduction
10X snRNA-seq and snATAC-seq was ran on 6 regions of the heart from 3 individuals. Here we explore the data from one donor (03231). Generally the data were good quality. We removed cells with small libraries (< 1000 UMI in RNA, < 3000 unique fragments in ATAC). We also removed doublets in ATAC-seq using ArchR's doublet removal scheme. For RNA-seq, we simply removed outlier libraries according to UMI count to account for doublets. We were left with 27,983 and 11,777 nuclei in RNA-seq and ATAC-seq.
Clustering
The dimensionality reduction and clustering was done outside of this notebook and the resulting objects were serialized/saved. Details as to how the data were processed are in scripts/.
suppressMessages(library(Seurat))
suppressMessages(library(ArchR))
suppressMessages(source('../scripts/analysis_utils.R'))
srna <- readRDS('../seurat/Heart_RNA_list_processed.rds')
srna <- srna$`03231`
satac <- suppressMessages(loadArchRProject('../ArchR_project_03231/', showLogo = F))We can visualize the six different regions:
palette <- setNames(brewer.pal(6, "Set2"), unique(srna$region))
p1 <- DimPlot(srna, label = F, group.by='region', cols = palette) + ggClean() + ggtitle('scRNA-seq')
p2 <- custom_archr_umap(archr_project = satac, group.by = 'regions', palette = palette, legend = F, label = F, pt.size = 0.05) + ggtitle('scATAC-seq')
p1 + p2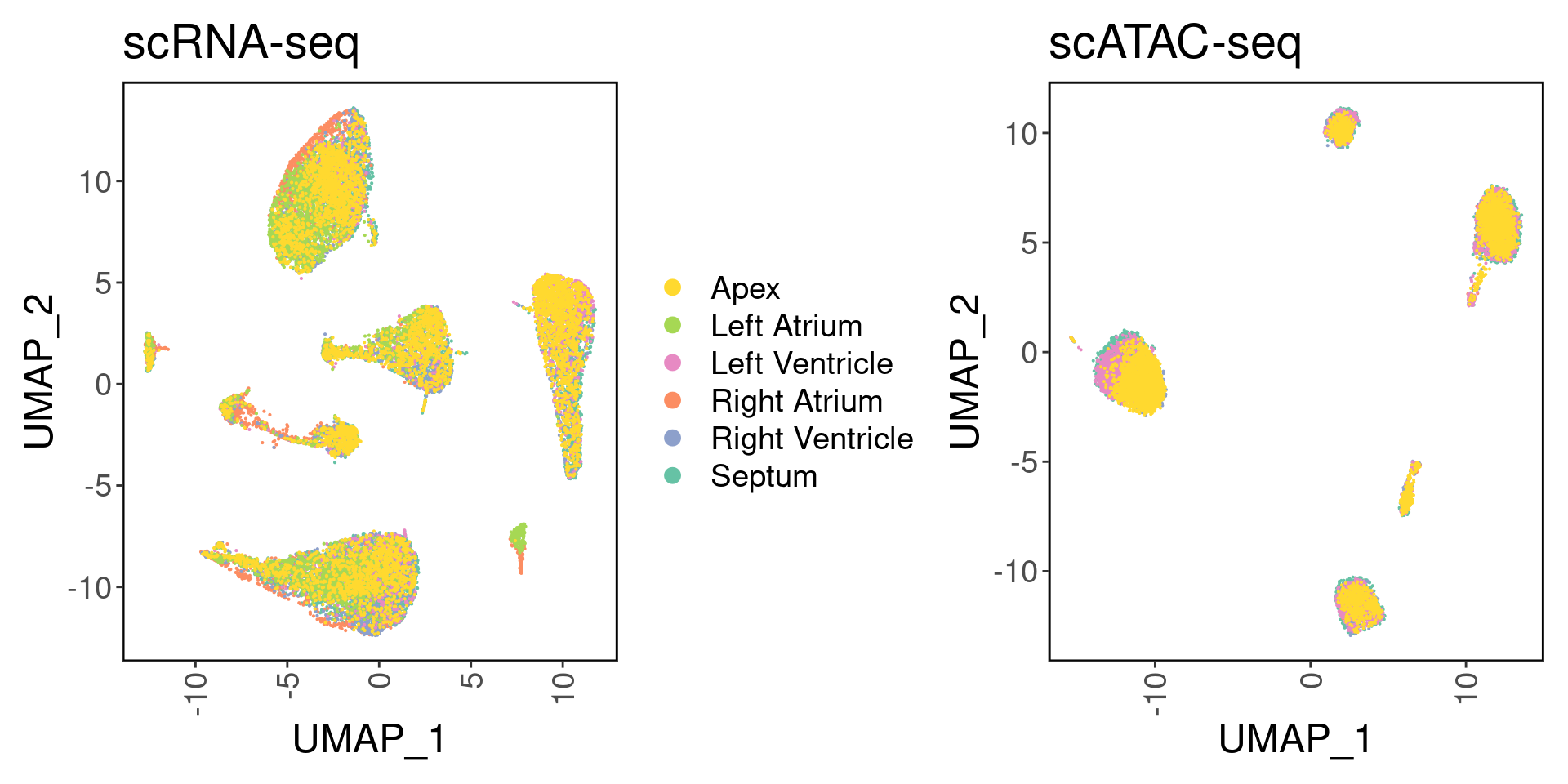
We can visualize the clustering results with the inferred cell types. I used the following markers to define cell types:
| marker | CellType |
|---|---|
| TTN/MYBPC3 | Cardiomyocytes |
| MYH7 | Vent. CM |
| NPPA | Atrial CM |
| RGS5/ABCC9 | Pericyte |
| MYH11/TAGLN | Smooth Muscle |
| DCN/PDGFRA | Fibroblast |
| PECAM1/VWF | Endothelial |
| PLP1 | Neuronal |
| CDA8/LCK | Lymphoid |
| CD14/FOLR2 | Myeloid |
palette <- setNames(brewer.pal(9, "Set3"), levels(Idents(srna)))
p1 <- DimPlot(srna, label = T, cols = palette) + ggClean() + NoLegend() + ggtitle('scRNA-seq')
p2 <- custom_archr_umap(archr_project = satac, group.by = 'CellTypes', palette = palette, legend = F, label = T) + ggtitle('scATAC-seq')
p1 + p2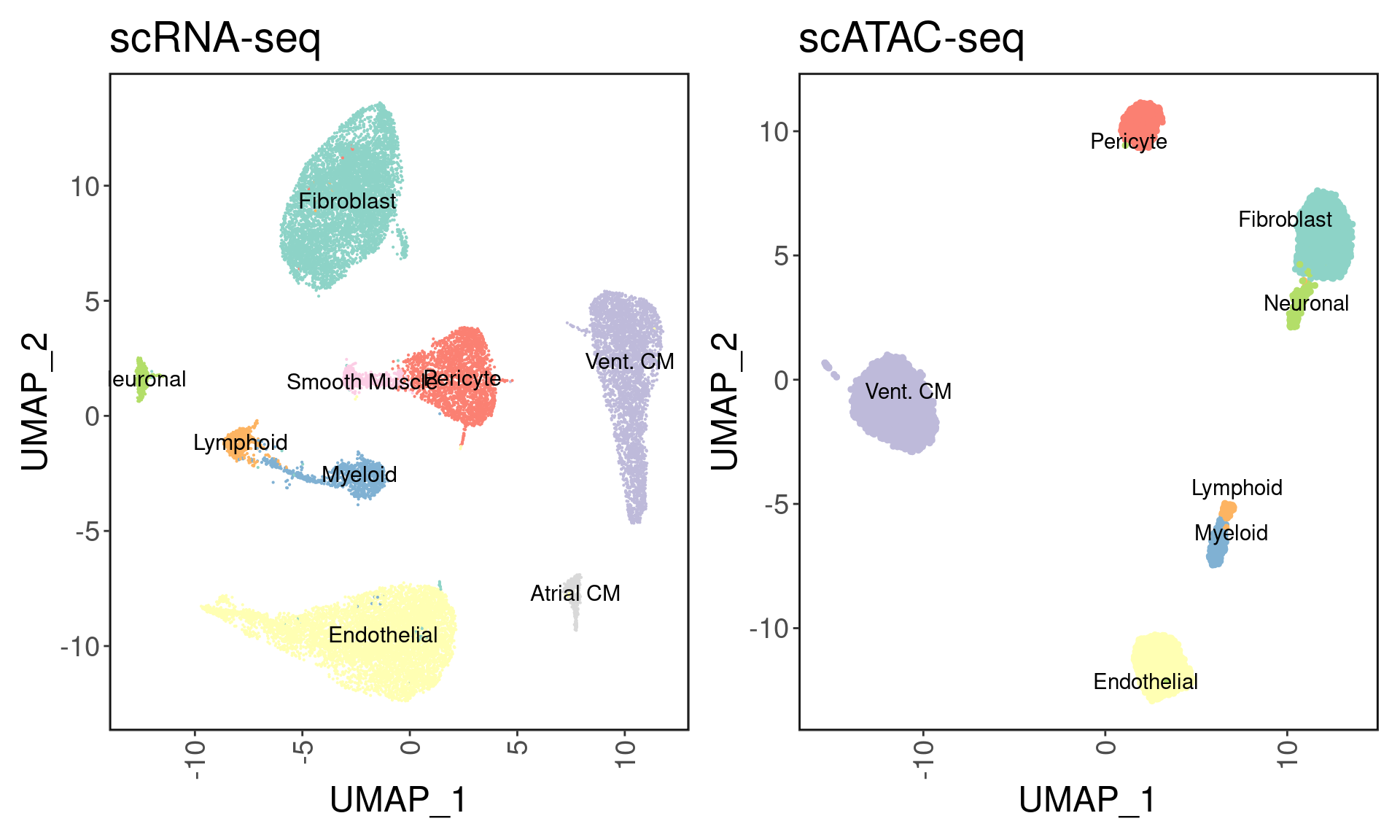
We recover 9 cell types in scRNA-seq and 7 in scATAC-seq. How do they compare in proportion?
As expected, cardiomyocytes are the dominant cell type in both assays.
freq <- table(Idents(srna))/length(Idents(srna))
p1 <- make_freq_plot(freq, palette = palette) + LegendOff() + ggtitle('scRNA-seq')
freq <- table(satac$CellTypes)/length(satac$CellTypes)
p2 <- make_freq_plot(freq, palette = palette) + LegendOff() + ggtitle('scATAC-seq')
p1 + p2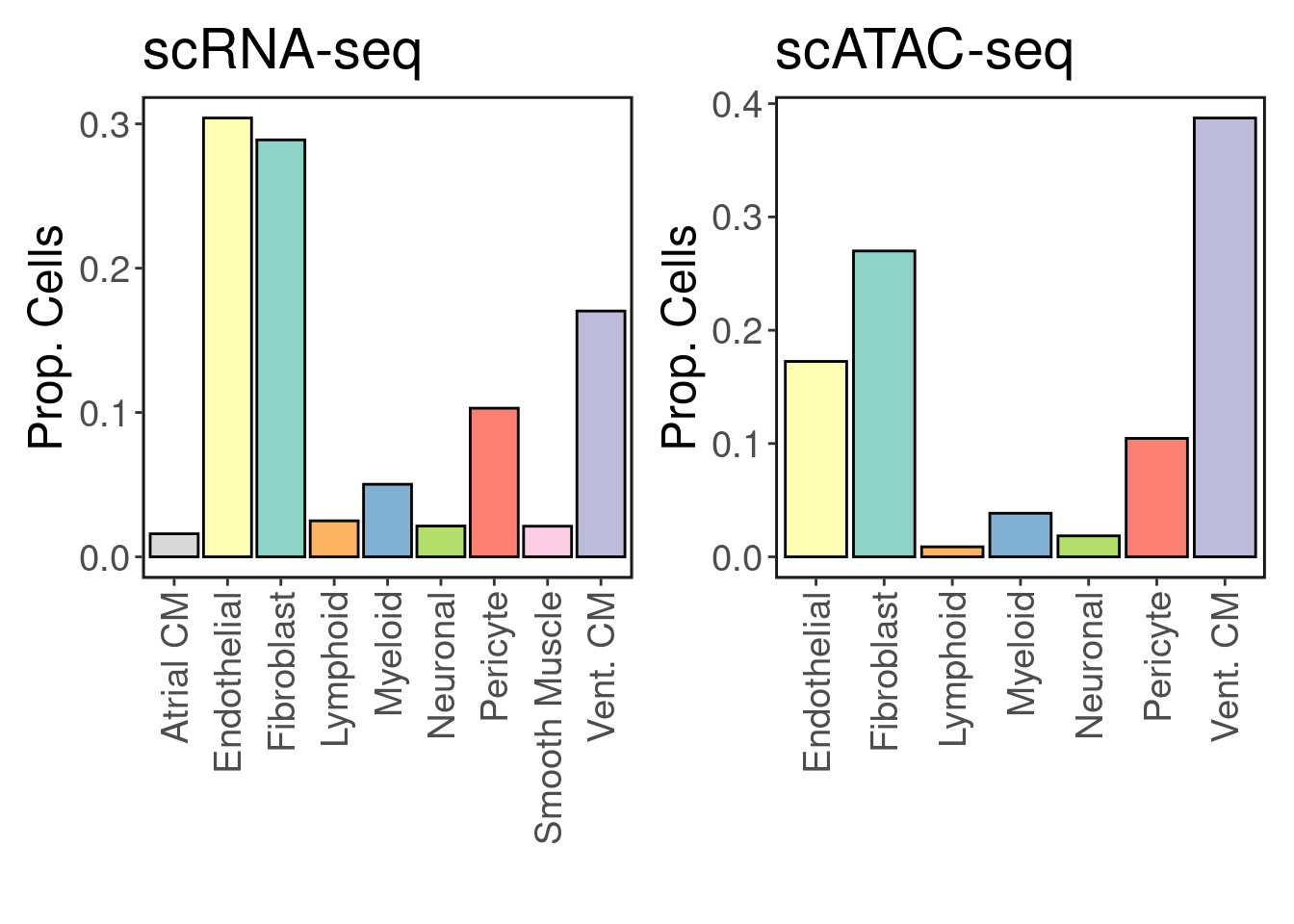
CRE discovery
Here we run macs2 on ATAC-seq aggregates of each cell-type to discover cell-type specific peaks. Below shows the number of peaks called in total and for each cell type. As expected, most peaks are not protein coding.
peak.info <- getPeakSet(satac)
peak.info.per <- peak.info@metadata$PeakCallSummary
pal <- c("Distal" = "#60BA64", "Exonic" = "#73C6FF", "Intronic" = "#620FA3", "Promoter" = "#FFC554")
ggplot(peak.info.per, aes(x=Group, y=Freq, fill=Var1)) + geom_bar(stat='identity') + labs(fill='') +
scale_fill_manual(values = pal) + ggClean() + ylab("Number of Peaks \n (10^3)")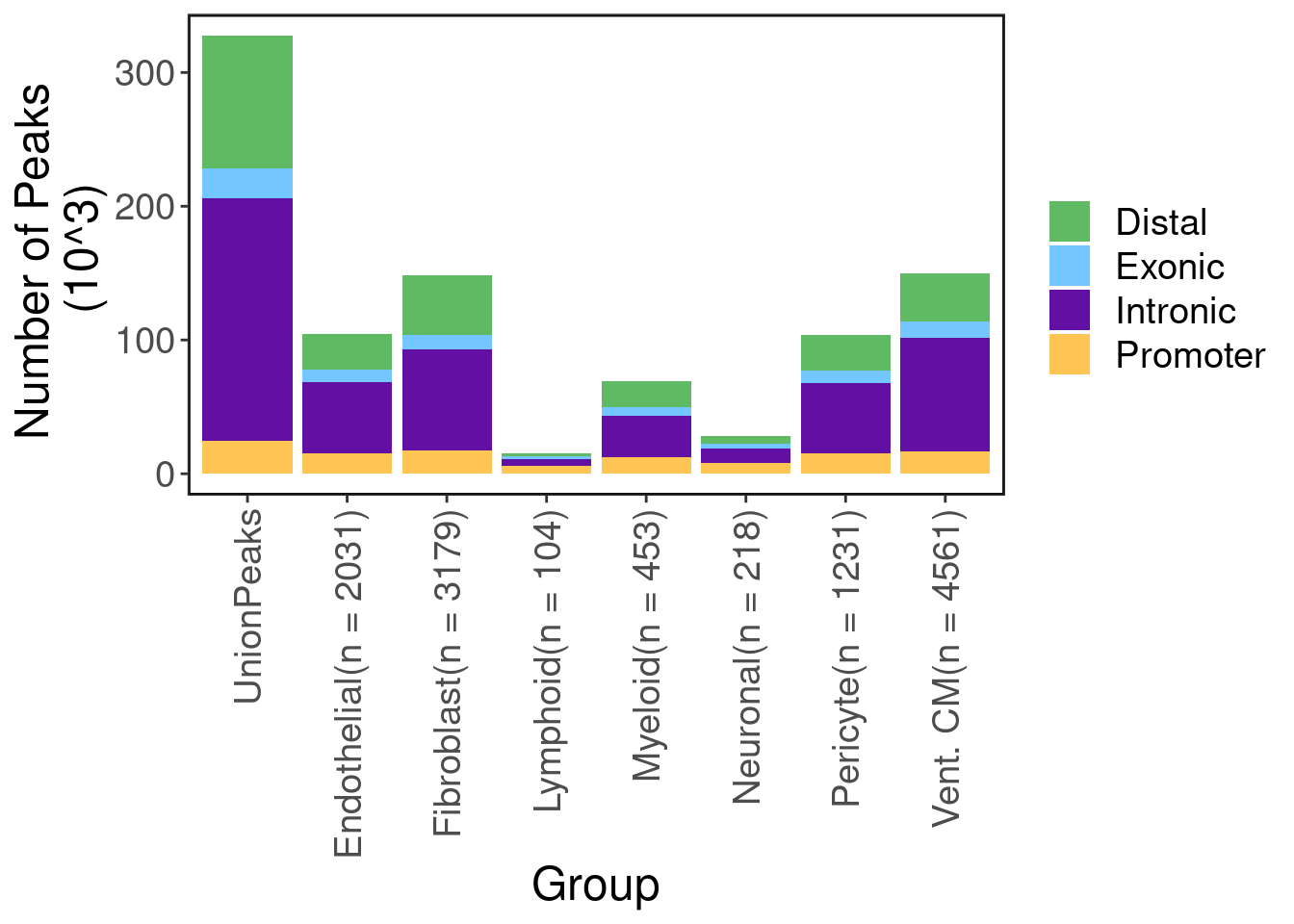
Based on the union set, we create a single-cell binary peak accessibility matrix and find the peaks that are differentially accessible. Below we plot the relative accessiblility across cells for differentially accessible peaks. We only plot peaks with FDR < 10% and log2 fold-change > 0.5. Based on this filtering, we have discovered 170,0529 regulatory elements.
markersPeaks <- readRDS('../ArchR_project_03231/PeakCalls/DA_markerPeaks.rds')
suppressMessages(plotMarkerHeatmap(seMarker = markersPeaks, cutOff = "FDR <= 0.1 & Log2FC >= 0.5", transpose = TRUE))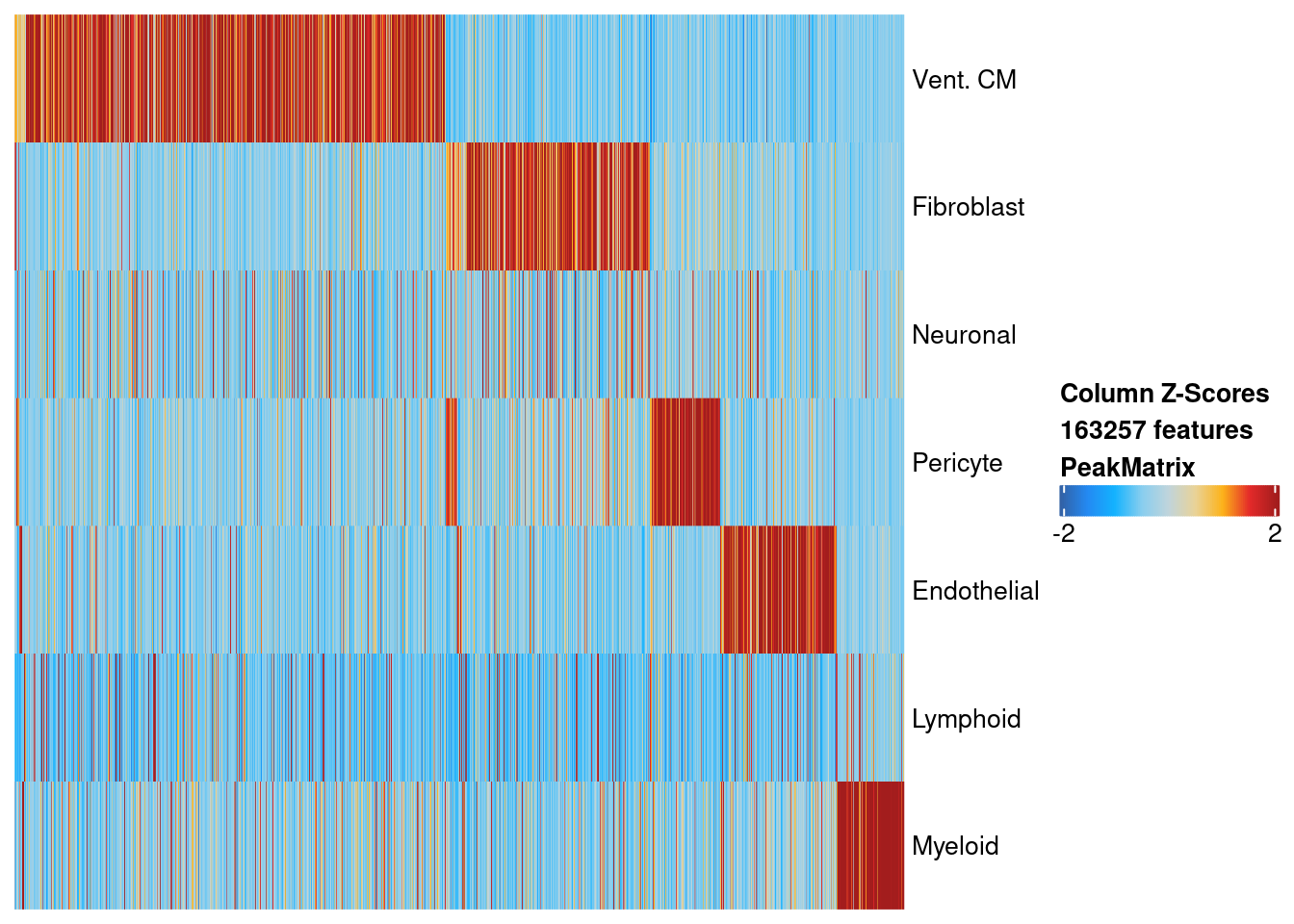
Exploration: GTEx enrichment
GTEx has eQTL data for n = 495 individuals for heart tissue (left ventricle.) We hypothesize that the cardiomyocyte (CM) regulatory elements are enriched with heart eQTLs. We use Torus to perform this enrichment analysis. We plot the log2 enrichment result below.
enrich.df <- read.delim('../eQTL_enrich/results/donor_03231_enrichment.txt', header=F, sep="")
ggplot(enrich.df, aes(x=V2, y=V1)) + geom_point() + geom_errorbar(aes(xmin=V3, xmax=V4), colour="black", width=.1) + ggClean() +
xlab('log2 Enrichment') + ylab('Cell Type') + xlim(c(0,2.5)) + geom_vline(xintercept = 0, col='red', linetype = "longdash")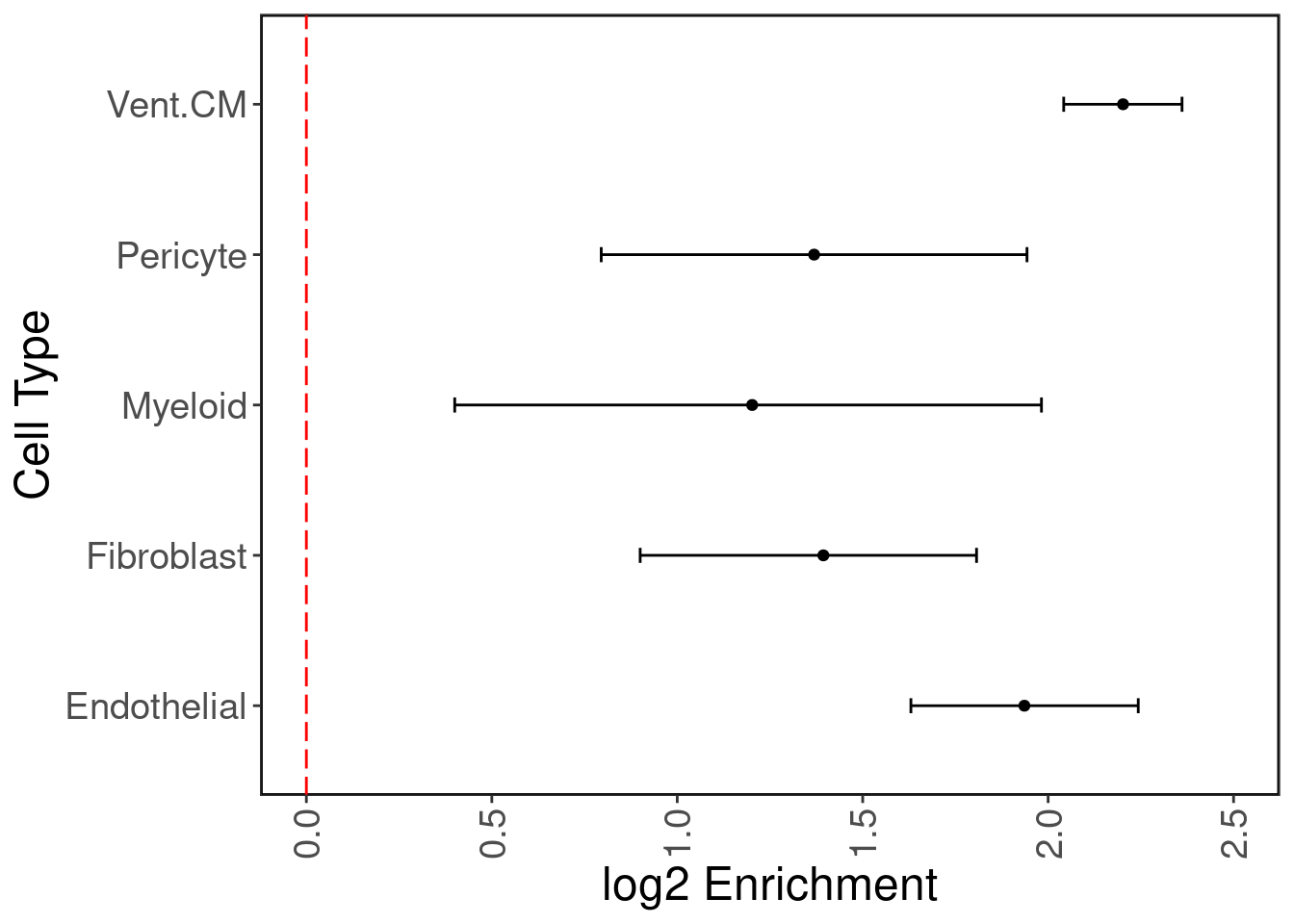
As expected, ventricular cardiomyocyte peaks are enriched with heart eQTLs, with endothelial peaks as second most enriched.